10 août 2023
Des chercheurs parviennent à renverser certaines manifestations de l'alzheimer dans un modèle animal de la maladie
Le maintien d'une pompe à ions située dans la membrane cellulaire des neurones permettrait de ralentir ou de renverser la pathologie

'%3e%3cpath%20d='M306.615%2079.694H144.011L892.476%201150.3h162.604ZM0%200h357.328l309.814%20450.883L1055.03%200h105.86L714.15%20519.295%201200%201226.37H842.672L515.493%20750.215%20105.866%201226.37H0l468.485-544.568Z'/%3e%3c/g%3e%3c/svg%3e)


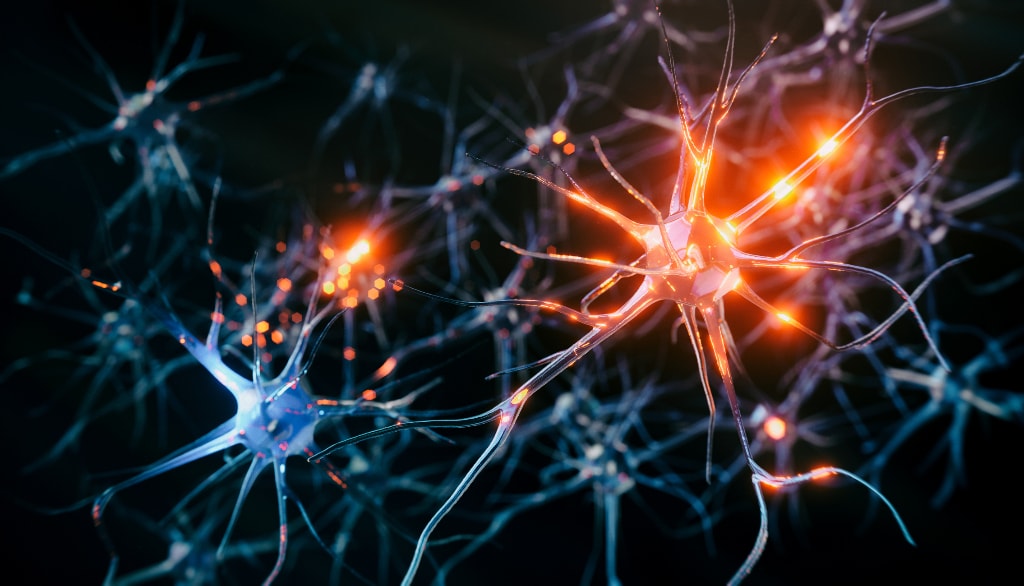
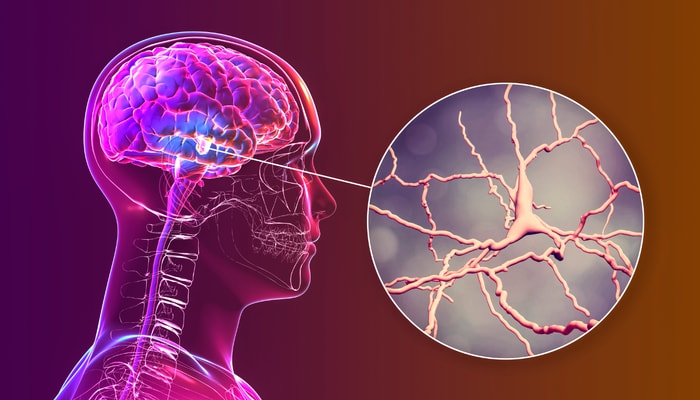
Lorsqu'il y a diminution de la molécule KCC2 dans la membrane cellulaire des neurones, leur équilibre ionique est perturbé et ils deviennent hyperactifs, comme le suggère cette illustration. Cette hyperactivité peut conduire à la mort des neurones.
— Getty Images/Koto Feja
Une équipe de chercheurs annonce, dans la revue Brain, qu'elle est parvenue à renverser certaines manifestations cognitives associées à l'alzheimer dans un modèle animal de la maladie. «Même si la démonstration reste à faire chez l'humain, nous croyons que le mécanisme que nous avons mis en lumière constitue une cible thérapeutique très intéressante parce qu'il ne se limite pas à ralentir la progression de la maladie, mais qu'il permet de restaurer en partie certaines fonctions cognitives», commente le responsable de l'étude, Yves De Koninck, professeur à la Faculté de médecine et chercheur au Centre de recherche CERVO de l'Université Laval.
Des études antérieures ont montré qu'avant même que les symptômes de l'alzheimer apparaissent, l'activité du cerveau est perturbée chez les gens qui vont développer la maladie. «Il y a une hyperactivité neuronale et une désorganisation des signaux dans le cerveau, explique le chercheur. Notre hypothèse est qu'un mécanisme qui régule l'activité neuronale, plus précisément celui qui est responsable de l'inhibition des signaux neuronaux, est perturbé.»
Le principal inhibiteur des signaux neuronaux dans le cerveau humain est le neurotransmetteur GABA. Il fonctionne en étroite collaboration avec un cotransporteur, le KCC2. Il s'agit d'une pompe à ions, située dans la membrane cellulaire, qui fait circuler les ions chlorures et les ions potassium entre l'intérieur et l'extérieur des neurones, rappelle le professeur De Koninck.
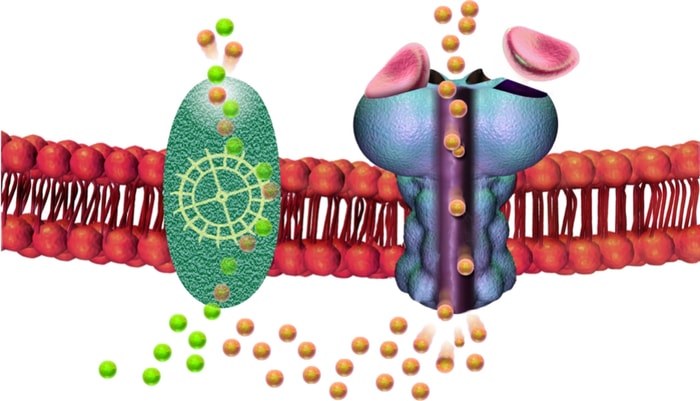
Le récepteur GABA (en bleu) laisse entrer les ions chlorures négatifs (en jaune) dans les neurones, ce qui inhibe la transmission de l'influx nerveux. Pour que cette inhibition se maintienne, il faut que des ions chlorures soient constamment expulsés de la cellule. Le cotransporteur KCC2 (en vert) se charge de cette tâche en pompant un ion chlorure négatif en même temps qu'un ion potassium positif (en vert pâle).
— Bruce Blauss/Sylvain Côté
«Lorsqu'il y a une perte de KCC2 dans la membrane cellulaire, le niveau d'ions chlorures augmente à l'intérieur des neurones, et l'inhibition médiée par GABA est perturbée, poursuit-il. Cela peut conduire à l'hyperactivité neuronale. Une étude a déjà montré que les niveaux de KCC2 étaient réduits dans le cerveau de personnes décédées qui avaient souffert d'alzheimer. C'est ce qui nous a donné l'idée d'examiner le rôle de KCC2 dans un modèle animal de la maladie d'Alzheimer.»
Pour ce faire, les chercheurs ont utilisé une lignée de souris qui expriment l'une des deux principales manifestations de l'alzheimer chez l'humain: la formation de plaques amyloïdes dans le cerveau. Comme prévu, ces plaques sont apparues dans le cerveau de ces souris et leur abondance a augmenté avec l'âge.
Les chercheurs ont constaté que, lorsque ces souris atteignaient l'âge de 4 mois, les niveaux de KCC2 diminuaient dans deux régions de leur cerveau. Ces deux régions, l'hippocampe et le cortex préfrontal, sont aussi affectées chez les personnes qui souffrent d'alzheimer. «Plus la perte de KCC2 était grande, plus les souris avaient de plaques amyloïdes», note le professeur De Koninck.
À la lumière de ces résultats, les chercheurs ont eu recours à une molécule mise au point dans leur laboratoire, le CLP290, un activateur de KCC2 qui prévient sa diminution. À court terme, l'administration de cette molécule à des souris qui avaient déjà des niveaux de KCC2 réduits a permis une amélioration de leur mémoire spatiale et de leurs comportements sociaux. À long terme, le CLP290 les a protégées contre une diminution des capacités cognitives et contre l'hyperactivité neuronale.
«Nos résultats n'impliquent pas que la perte de KCC2 cause l'alzheimer, insiste le professeur De Koninck. Par contre, elle semble entraîner un déséquilibre ionique conduisant à une hyperactivité neuronale pouvant mener à la mort des neurones. Cela suggère qu'en prévenant la perte de KCC2, on pourrait ralentir et peut-être même renverser certaines manifestations de la maladie.»
— Yves De Koninck
Pour différentes raisons, le CLP290 ne peut être utilisé chez l'humain. L'équipe du professeur De Koninck est en quête d'autres molécules activatrices de KCC2 qui seraient bien tolérées par les personnes souffrant d'alzheimer. «Nous avons développé de nouvelles molécules qui sont présentement en évaluation dans notre laboratoire. Parallèlement à ces recherches, nous testons des médicaments qui sont utilisés à des fins autres que l'alzheimer chez l'humain afin d'en évaluer les effets sur le KCC2. Le repositionnement d'un médicament existant accélérerait les travaux portant sur cette nouvelle avenue thérapeutique», souligne le chercheur.
Les autres signataires de l'étude parue dans Brain sont Iason Keramidis, Julien Bourbonnais, Feng Wang, Dominique Isabel, Marie-Eve Paquet, Romain Sansonetti, Annie Barbeau, Lionel Froux et Antoine Godin, de l'Université Laval, et Brendan McAllister, Edris Rezaei, Phil Degagne, Mojtaba Nazari, Samsoon Inayat et Majid Mohajerani, de l'Université de Lethbridge.